
O alinhamento múltiplo sequência
Fundo para as escolas Wikipédia
Crianças SOS oferecem um download completo desta seleção para as escolas para uso em escolas intranets. patrocínio SOS Criança é legal!
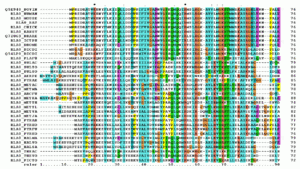
Um alinhamento de sequências múltiplas (MSA) é um alinhamento da sequência de três ou mais sequências biológicos, em geral, proteínas , ADN , ou ARN. Em geral, o conjunto de entrada de seqüências de consulta são assumidos ter uma evolutiva relacionamento pelo qual eles compartilham uma linhagem e são descendentes de um ancestral comum. A partir do MSA, a sequência resultante a homologia pode ser inferida e análise filogenética pode ser conduzido para avaliar origens evolutivas compartilhadas das sequências. Representações visuais do alinhamento como na imagem à direita ilustram eventos de mutação, tais como mutações pontuais (único aminoácido ou alterações de nucleótidos) que aparecem como personagens diferentes em uma única coluna alinhamento e inserção ou deleção mutações (ou indels) que aparecem como lacunas em uma ou mais das sequências do alinhamento. Alinhamento múltiplo de sequências é muitas vezes utilizado para avaliar sequência de conservação domínios proteicos, terciária e estruturas secundárias, e ainda aminoácidos individuais ou nucleótidos.
O alinhamento múltiplo seqüência também se refere ao processo de alinhar um conjunto tão sequência. Porque três ou mais sequências de comprimento biologicamente relevante pode ser difícil e são quase sempre à mão para alinhar demorado, computacionais algoritmos são utilizados para produzir e analisar os alinhamentos. MSAs exigem metodologias mais sofisticadas do que o alinhamento aos pares , porque eles são computacionalmente mais complexo para produzir. A maioria dos programas de alinhamento de seqüência múltipla usar métodos heurísticos, em vez de otimização global porque identificar o alinhamento ótimo entre mais do que algumas seqüências de comprimento moderado é proibitivamente dispendiosa.
A programação dinâmica e complexidade computacional
O método mais directo para a produção de um MSA utiliza o técnica de programação dinâmica para identificar a solução globalmente alinhamento ideal. Para proteínas, este método envolve geralmente dois conjuntos de parâmetros: uma e uma penalidade de lacuna matriz de substituição atribuindo pontuações ou probabilidades para o alinhamento de cada par possível de aminoácidos com base na similaridade das propriedades químicas dos ácidos aminados 'e a probabilidade evolutiva da mutação. Para sequências de nucleótidos uma matriz de substituição pode ser usado, mas uma vez que existem apenas quatro caracteres possíveis padrão por sequência e os nucleótidos individuais não diferem muito tipicamente na probabilidade de substituição, os parâmetros para sequências de ADN e de ARN consistem geralmente de uma penalidade de intervalo, uma positiva nota pelo caráter jogos, e uma pontuação negativa para descasamentos.
Para n sequências individuais, o método requer a construção do equivalente n-dimensional da matriz formada em programação dinâmica pares padrão. O espaço de busca aumenta exponencialmente com o aumento, assim, n e é também fortemente dependente do comprimento de sequência. Para encontrar o ótimo global para n sequências desta forma foi demonstrado ser um Problema NP-completo. Métodos para reduzir o espaço de busca em primeiro lugar a execução da programação dinâmica aos pares em cada par de sequências no conjunto de consulta e busca apenas o espaço solução perto estes resultados (efetivamente encontrar a interseção entre caminhos locais imediatamente ao redor de cada solução ideal par a par) tornar a técnica de programação dinâmica mais eficiente. O chamado "soma de pares de" método foi implementado no pacote de software MSA, mas ainda é impraticável para muitas aplicações que exigem que o MSA alinhamento simultânea de dezenas ou mesmo algumas centenas de sequências. Métodos de programação dinâmica são agora usados apenas quando é necessário um alinhamento de um pequeno número de sequências extremamente elevada qualidade, e como um aferição padrão na avaliação de técnicas heurísticas novos ou refinados.
Construção alinhamento progressivo
Um método de realizar uma pesquisa de alinhamento heurística é a técnica progressiva (também conhecido como o método hierárquica ou em árvore) que acumula uma MSA final pela primeira realização de uma série de alinhamentos entre pares de sequências sucessivamente menos estreitamente relacionados. Tais métodos começam por alinhamento das duas sequências mais proximamente relacionadas em primeiro lugar e, em seguida, alinhando sucessivamente o seguinte sequência mais intimamente relacionado na consulta para definir o alinhamento produzido no passo anterior. O par "mais relacionado" inicial é determinada por um eficiente método de agrupamento tal como vizinho a se associar com base em uma pesquisa heurística simples da consulta definida com uma ferramenta como FASTA. Técnicas avançadas, portanto, construir automaticamente uma árvore filogenética, bem como um alinhamento.
Uma grande limitação de métodos progressivos é a sua grande dependência do atribuição inicial de parentesco e sobre a qualidade do alinhamento inicial. Os métodos são, portanto, sensíveis, bem como para a distribuição de sequências no conjunto de consulta; o desempenho melhora quando parentesco entre as seqüências de consulta é um inclinação relativamente suave, em vez de aglomerados distantes separados. O desempenho também degrada significativamente quando todas as sequências no conjunto são bastante distante relacionados, porque imprecisões no alinhamento inicial são, em seguida, mais provável. A maioria dos métodos modernos progressivas modificar a sua função de pontuação com uma função de ponderação secundário que atribui factores de escala para membros individuais da consulta definir de uma forma não linear com base na sua distância filogenética de seus vizinhos mais próximos. Escolha criteriosa de ponderação pode auxiliar na avaliação de parentesco e mitigar os efeitos de alinhamentos iniciais relativamente pobres no início da progressão.
Métodos de alinhamento progressivo são eficientes o suficiente para implementar em larga escala para muitas sequências e muitas vezes são executados em servidores web publicamente acessíveis para que os usuários não precisam localmente instalar as aplicações de interesse. Um método de alinhamento progressivo muito popular é o Clustal família, especialmente a variante ponderada ClustalW aos quais o acesso é fornecido por um grande número de portais web, incluindo GenomeNet, EBI, e EMBNet. Diferentes portais ou implementações podem variar em interface com o usuário e fazer diferentes parâmetros acessíveis para o usuário. Clustal é amplamente utilizado para a construção árvore filogenética e como entrada para a previsão da estrutura de proteínas por modelagem por homologia.
Outro método de alinhamento progressivo comum chamada T-Coffee é mais lento que Clustal e seus derivados, mas geralmente produz alinhamentos mais precisos para conjuntos de seqüência de parentesco distante. T-Coffee calcula alinhamentos de pares combinando o alinhamento direto do par com alinhamentos indiretos que alinha cada seqüência do par de uma terceira sequência. Ele usa a saída de Clustal, bem como um outro programa de alinhamento local LALIGN, que encontra múltiplas regiões de alinhamento local entre duas sequências. O alinhamento resultante e árvore filogenética são utilizados como um guia para a produção de novos e mais precisos factores de ponderação.
Porque os métodos progressivos são heurísticas que não são garantidos para convergir para um ótimo global, a qualidade de alinhamento pode ser difícil de avaliar e seu verdadeiro significado biológico pode ser obscuro. Um método semi-progressive muito recente que melhora a qualidade de alinhamento e não usa uma heurística com perdas enquanto ainda estiver executando em tempo polinomial foi implementado no programa PSAlign.
Métodos iterativos
Um conjunto de métodos para produzir as AFM enquanto reduz os erros inerentes métodos progressivos são classificados como "iterativo", porque eles funcionam de forma semelhante aos métodos progressivos mas realinhar repetidamente as sequências iniciais, bem como adicionar novas sequências para a crescente MSA. Uma razão métodos progressivas são tão fortemente dependente de um alinhamento inicial de alta qualidade é o facto de que estes alinhamentos são sempre incorporada no resultado final - ou seja, uma vez que uma sequência tenha sido alinhada para o MSA, o alinhamento não é considerado ainda mais. Esta aproximação melhora a eficiência de custo na precisão. Em contrapartida, métodos iterativos pode voltar para alinhamentos de pares previamente calculados ou sub-MSAs incorporando sub-grupos da seqüência de consulta como um meio de otimizar um general função objetivo tais como encontrar uma pontuação de alinhamento de alta qualidade.
Uma variedade de sutilmente diferentes métodos de iteração foram implementadas e disponibilizadas em pacotes de software; análises e comparações têm sido úteis, mas geralmente abster-se de escolher um "melhor" técnica. O pacote de software Prrn / PRRP utiliza um algoritmo hill-escalando para otimizar sua pontuação de alinhamento MSA e iterativa corrige ambos os pesos de alinhamento e ou regiões "Gappy" localmente divergentes do crescimento MSA. PRRP executa melhor quando refinando um alinhamento previamente construído por um método mais rápido.
Outro programa iterativo, DIALIGN, tem uma abordagem incomum de se concentrar estritamente em alinhamentos locais entre os sub-segmentos ou motivos de sequência sem introduzir uma penalidade de intervalo. O alinhamento dos motivos individuais é então conseguida com uma representação de matriz semelhante a uma trama de matriz de pontos num alinhamento par a par. Um método alternativo que utiliza rápidos alinhamentos locais como pontos de ancoragem ou "sementes" para um procedimento de alinhamento global mais lenta é implementado no CHAOS / DIALIGN suite.
Um terceiro método baseado em iteração popular chamado MÚSCULO (alinhamento múltiplo seqüência de log-expectativa) melhora em métodos progressivos com uma medida mais exata distância para avaliar o relacionamento de duas seqüências. A medida de distância é atualizado entre os estágios de iteração (embora, na sua forma original, músculo continha apenas 2-3 iterações, dependendo se o refinamento foi ativado).
Modelos ocultos de Markov
Modelos ocultos de Markov são modelos probabilísticos que pode atribuir probabilidades para todas as combinações possíveis de lacunas, jogos e incompatibilidades para determinar a mais provável MSA ou conjunto de possíveis MSAs. HMMs pode produzir uma única saída maior pontuação, mas também pode gerar uma família de alinhamentos possíveis que podem então ser avaliadas para significância biológica. Porque HMMs são probabilísticas, eles não produzem a mesma solução cada vez que são executados no mesmo conjunto de dados; assim, eles não pode ser garantida a convergir para um alinhamento óptimo. HMMs pode produzir alinhamentos globais e locais. Embora os métodos baseados em HMM tem sido desenvolvido relativamente pouco tempo, que oferecem melhorias significativas na velocidade computacional, especialmente para sequências que contêm regiões que se sobrepõem.
Típico baseado em HMM métodos de trabalho, o que representa um MSA como uma forma de grafo acíclico dirigido conhecido como um gráfico de ordem parcial, que consiste de uma série de nós que representam possíveis entradas nas colunas de um MSA. Nesta representação, uma coluna que é absolutamente conservado (isto é, que todos as sequências de partes da MSA um carácter particular a uma posição em particular) é codificada como um único nó com o maior número de ligações de saída uma vez que existem possíveis caracteres na coluna seguinte de o alinhamento. Nos termos de um modelo típico oculto de Markov, os estados observados são as colunas de alinhamento individuais e os estados "escondidos" representam a sequência ancestral presumida a partir do qual as sequências no conjunto de consulta são a hipótese de ter descido. Uma variante de busca eficiente de o método de programação dinâmico, conhecido como o Algoritmo de Viterbi, é geralmente utilizado para alinhar sucessivamente crescente MSA para a próxima sequência na consulta configurada para produzir um novo MSA. Isso é diferente de métodos de alinhamento progressivo porque o alinhamento de sequências anteriores é atualizada a cada nova adição sequência. No entanto, como métodos progressivos, esta técnica pode ser influenciado pela ordem em que as sequências no conjunto de consulta estão integrados no alinhamento, especialmente quando as sequências estão distantemente relacionados.
Vários programas de software estão disponíveis em que as variantes de métodos baseados em HMM têm sido implementadas e que são conhecidos por sua escalabilidade e eficiência, embora corretamente, usando um método HMM é mais complexa do que usando métodos progressivos mais comuns. O mais simples é POA (Partial-Order Alinhamento); um método semelhante, mas mais generalizada é implementado no pacote SAM (Sequência Alinhamento e Modeling System). SAM tem sido usado como uma fonte de alinhamentos de a previsão da estrutura de proteínas para participar no CASP experimento previsão da estrutura e para desenvolver uma base de dados de proteínas previstas nas leveduras espécies De S. cerevisiae. HMM métodos também podem ser usados para a pesquisa de banco de dados com HMMER.
Algoritmos genéticos e recozimento simulado
Técnicas padrão de otimização em ciência da computação - ambos os quais foram inspirados por, mas não se reproduzem diretamente, processos físicos - também têm sido utilizados na tentativa de produzir de forma mais eficiente as AFM qualidade. Uma tal técnica, algoritmos genéticos, tem sido utilizada para a produção de MSA em uma tentativa de simular amplamente o processo evolutivo a hipótese de que deu origem à divergência no conjunto de consulta. O método funciona através da quebra de uma série de possíveis AFM em fragmentos e repetidamente rearranjar os fragmentos, com a introdução de intervalos nas posições diferentes. Um general função objetivo é otimizado durante a simulação, a maioria geralmente a "soma de pares de" função de maximização introduzido em métodos MSA dinâmicas baseadas em programação. Uma técnica para sequências de proteínas foi implementado no programa de software SAGA (Sequência Alinhamento pelo Algoritmo Genético) e seu equivalente em RNA é chamado RAGA.
A técnica de recozimento simulado, pelo que um MSA existente produzido por um outro método é refinado por uma série de rearranjos concebidos para encontrar as regiões mais ideal do espaço de alinhamento do que aquela que o alinhamento de entrada já ocupa. Como o método de algoritmos genéticos, recozimento simulado maximiza uma função objetivo como a função de soma de pares. Recozimento simulado usa uma metafórica "fator de temperatura" que determina a taxa em que os rearranjos e proceder a probabilidade de cada rearranjo; típicos períodos suplentes uso de altas taxas de rearranjo com relativamente baixa probabilidade (para explorar regiões mais distantes do espaço alinhamento) com períodos de taxas mais baixas e mais altas probabilidades para explorar mais profundamente mínimos locais perto das regiões recentemente "colonizados". Esta abordagem tem sido implementada no programa Msasa (Multiple alinhamento de sequências de Simulated Annealing).
Constatação Motif

Descoberta do motivo, também conhecido como análise de perfil, é um método de localização motivos de sequência de AFM globais que é ao mesmo tempo um meio de produzir um MSA melhor e um meio de produzir uma matriz de pontuação para o uso em outras sequências para pesquisar motivos semelhantes. Uma variedade de métodos para isolar os motivos têm sido desenvolvidos, mas todos são baseados na identificação de padrões curto altamente conservadas dentro do alinhamento maior e construção de uma matriz semelhante a uma matriz de substituição que reflecte a composição de aminoácidos ou de nucleótidos de cada posição no motivo putativo . O alinhamento pode ser então refinada usando estas matrizes. Na análise do perfil padrão, a matriz inclui entradas para cada personagem possível, bem como entradas para as lacunas. Alternativamente, encontrando-algoritmos estatísticos padrão pode identificar motivos como um precursor para um MSA, em vez de como uma derivação. Em muitos casos quando o conjunto de consulta contém apenas um pequeno número de sequências ou sequências contém apenas altamente relacionados pseudocounts são adicionados a normalizar a distribuição reflecte-se na matriz de pontuação. Em particular, esta entrada corrige-probabilidade zero na matriz para valores que são pequenas, mas diferente de zero.
Análise blocos é um método de encontrar motivo que restringe motivos para regiões ungapped no alinhamento. Os blocos podem ser gerados a partir de um MSA ou podem ser extraídos a partir de sequências não alinhadas utilizando um conjunto pré-calculado de motivos comuns previamente gerado a partir de famílias de genes conhecidos. Bloco de pontuação geralmente baseia-se no espaçamento dos caracteres de alta frequência, em vez de sobre o cálculo de uma matriz de substituição explícita. O BLOCOS servidor fornece um método interativo para localizar tais motivos em sequências desalinhadas.
Estatística de correspondência de padrões tem sido implementado usando tanto o algoritmo de Maximização da Expectação eo Amostrador de Gibbs. Uma das ferramentas de apuramento motivo mais comuns, conhecidos como meme, usa maximização expectativa e métodos ocultos de Markov para gerar motivos que são então utilizados como ferramentas de pesquisa por seu companheiro MAST na suíte combinada MEME / MAST.