
Reacción aldólica
Antecedentes de las escuelas de Wikipedia
SOS ofrecen una descarga completa de esta selección de escuelas para su uso en escuelas intranets. Una buena manera de ayudar a otros niños es mediante el patrocinio de un niño
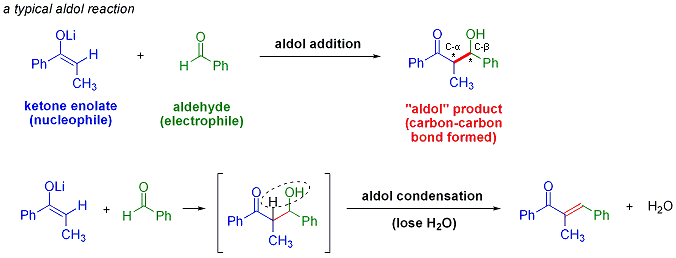
La reacción aldólica es un importante carbono-carbono formación de enlaces de reacción en química orgánica . En su forma habitual, implica el adición nucleofílica de un cetona enolato a una aldehído para formar una cetona β-hidroxi, o "aldol" (ald ehyde + alcoh ol), una unidad estructural que se encuentra en muchas moléculas de origen natural y productos farmacéuticos. A veces, el producto de adición aldólica pierde una molécula de agua durante la reacción para formar una α, β-insaturado cetona. Esto se llama una condensación aldólica. La reacción aldólica se descubrió de forma independiente por Charles-Adolphe Wurtz y por Alexander Borodin Porfyrevich en 1872. Borodin observó la dimerización aldólica de 3-hidroxibutanal de acetaldehído en condiciones ácidas. La reacción aldólica se utiliza ampliamente en la producción a gran escala de productos químicos básicos, tales como pentaeritritol y en el industria farmacéutica para la síntesis de fármacos ópticamente puros. Por ejemplo, la ruta inicial de Pfizer para el Lipitor de drogas enfermedad cardíaca (INN: atorvastatina), aprobado en 1996, empleó dos reacciones aldólicas, permitiendo el acceso a cantidades multigramo escala de la droga.
El motivo estructural aldólica es especialmente común en policétidos, una clase de productos naturales de los que se derivan muchos productos farmacéuticos, incluyendo el potente inmunosupresor FK506, la antibióticos de tetraciclina, y el agente antifúngico anfotericina B. Una amplia investigación sobre la reacción aldólica ha producido métodos altamente eficientes que permiten el reto de otro modo la síntesis de muchos policétidos en el laboratorio. Esto es importante porque muchos policétidos, junto con otras moléculas biológicamente activas, se producen naturalmente en cantidades pequeñas poco práctica para la investigación adicional. La síntesis de muchos de estos compuestos, una vez considerado casi imposible, ahora se puede realizar de forma rutinaria en la escala de laboratorio, y se está acercando a la viabilidad económica a una escala mayor en algunos casos, tales como el agente anti-tumor de gran actividad discodermolida. En la bioquímica , la reacción aldólica es uno de los pasos clave de glucólisis, donde es catalizada por enzimas llamadas aldolasas.
La reacción aldólica es particularmente valioso en síntesis orgánica, ya que produce productos con dos nuevos centros estereogénicos (en la α- y β-carbono del aducto de aldol, marcada con asteriscos en el esquema anterior). Los métodos modernos, que se describen a continuación, permiten ahora la configuración relativa y absoluta de estos centros para ser controlado. Esto es de particular importancia cuando la síntesis de productos farmacéuticos, ya que las moléculas con la misma conectividad estructural pero diferente estereoquímica menudo tienen muy diferentes propiedades químicas y biológicas.
Una variedad de nucleófilos se puede emplear en la reacción aldólica, incluyendo el enoles, enolatos, y enol éteres de cetonas, aldehídos, y otros muchos compuestos de carbonilo. La socio electrofílico es generalmente un aldehído, aunque muchas variaciones, tales como la Reacción de Mannich, existir. Cuando el nucleófilo y electrófilo son diferentes (el caso habitual), la reacción se llama una reacción aldólica cruzada (en oposición a dímeros formados en una dimerización aldólica).

Una solución de diisopropilamida de litio (LDA) en tetrahidrofurano (THF) (en el matraz a la derecha) se añadió a una solución de propionato de terc-butilo en el matraz a la izquierda, formando su enolato de litio. Un aldehído se puede añadir para iniciar una reacción de adición aldólica.
Ambos frascos se sumergen en un baño de hielo seco / acetona baño de enfriamiento (-78 ° C) la temperatura de la que está siendo monitoreado por un termopar (el cable de la izquierda).
Mecanismos
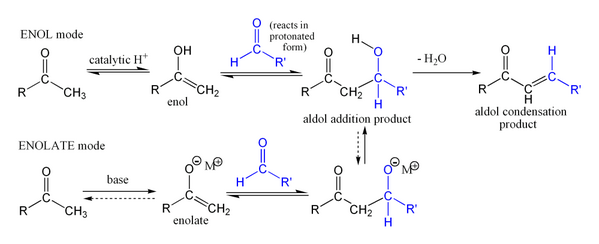
La reacción aldólica puede proceder a través de dos mecanismos fundamentalmente diferentes. Los compuestos de carbonilo, tales como aldehídos y cetonas, pueden ser convertidos a enoles o éteres de enol. Estos compuestos, siendo nucleófila en el α-carbono, puede atacar carbonilos protonadas especialmente reactivos, tales como aldehídos protonados. Este es el "mecanismo de enol". Los compuestos de carbonilo, siendo ácidos de carbono, también pueden ser desprotonados para formar enolatos, que son mucho más nucleófilo que enoles o éteres de enol y pueden atacar directamente electrófilos. El electrófilo usual es un aldehído, ya que las cetonas son mucho menos reactivos. Este es el "mecanismo enolato".
Si las condiciones son especialmente duras (por ejemplo, NaOMe, MeOH, reflujo), puede producirse condensación, pero esto generalmente pueden evitarse con reactivos suaves y temperaturas bajas (por ejemplo, LDA (una base fuerte), THF, -78 ° C). Aunque la adición aldólica generalmente procede a casi terminada, la reacción no es irreversible, ya que el tratamiento de aductos de aldol con bases fuertes generalmente induce la escisión retro-aldólica (da los materiales de partida). Condensaciones aldólicas son irreversibles.
Mecanismo de Enol
Cuando se utiliza un catalizador ácido, el paso inicial en el mecanismo de reacción implica catalizada por ácido tautomerización del compuesto de carbonilo a la enol. El ácido también sirve para activar el grupo carbonilo de otra molécula por protonación, haciéndolo altamente electrofílico. El enol es nucleófila en la α-carbono, permitiendo que se ataca el compuesto de carbonilo protonado, que conduce a la aldol después de desprotonación. Esto por lo general deshidrata para dar el compuesto de carbonilo insaturado. El esquema muestra una auto-condensación catalizada por ácido típico de un aldehído.
Aldólica catalizada por ácido mecanismo

Deshidratación catalizada por ácido

Mecanismo enolato
Si el catalizador es una base moderada tal como hidróxido de litio o una alcóxido, la reacción aldólica se produce a través de un ataque nucleofílico por el estabilizado por resonancia enolato en el grupo carbonilo de otra molécula. El producto es el sal alcóxido del producto aldol. El aldol en sí está formada a continuación, y puede a continuación someterse a deshidratación para dar el compuesto de carbonilo insaturado. El esquema muestra un mecanismo simple para la reacción aldólica catalizada por base de un aldehído consigo mismo.
Base catalizada reacción aldólica (que se muestra usando - OCH 3 como base)

Base deshidratación catalizada (escrito a veces como un solo paso)

Aunque sólo se requiere una cantidad catalítica de la base, en algunos casos, el procedimiento más habitual es utilizar una cantidad estequiométrica de una base fuerte tal como LDA o NaHMDS. En este caso, la formación de enolato es irreversible, y el producto aldol no se forma hasta que el alcóxido de metal del producto aldol se protona en una etapa de estudio diagnóstico separado.
Modelo de Zimmerman-Traxler
Se sabe que las formas más refinadas de mecanismo. En 1957, Zimmerman y Traxler propuesto que algunas reacciones aldólicas tienen "estado de transición de seis miembros s tiene una conformación de silla. "Esto que hoy se conoce como el modelo de Zimmerman-Traxler. E-enolatos dan lugar a productos contra, mientras que Z-enolatos dan lugar a productos syn. Los factores que controlan la selectividad son la preferencia para la colocación de los sustituyentes en el ecuador de seis miembros estados de transición y la evitación de interacciones syn-pentano, respectivamente. E y Z se refieren a la relación estereoquímica cis-trans entre el oxígeno enolato teniendo el contraión positivo y el grupo de prioridad más alta en el carbono alfa. En realidad, sólo algunos metales tales como litio y boro seguir de forma fiable el modelo de Zimmerman-Traxler. Así, en algunos casos, la resultado estereoquímico de la reacción puede ser impredecible.

Control en la reacción aldólica
El problema
El problema de "control" en la adición aldólica se demuestra mejor con un ejemplo. Considere el resultado de esta reacción hipotética:
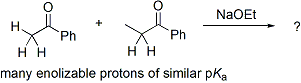
En esta reacción, dos cetonas asimétricas se condensan usando etóxido de sodio. La basicidad de etóxido de sodio es tal que no puede desprotonar totalmente cualquiera de las cetonas, pero puede producir pequeñas cantidades de enolato de sodio de ambos cetonas. Efectivamente, esto significa que además de ser potenciales electrófilos aldólicas, ambos cetonas también pueden actuar como nucleófilos a través de su enolato de sodio. Dos electrófilos y dos nucleófilos entonces potencialmente resulta en cuatro productos posibles:
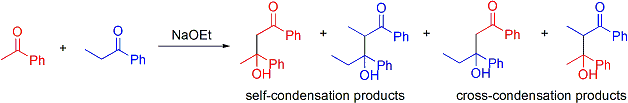
Por lo tanto, si se desea obtener sólo uno de los productos cruzados, entonces uno debe "controlar" la adición aldólica.
Acidez
Si un socio es considerablemente más ácido que el otro, entonces el control puede ser automático. El protón más ácido es abstraído por la base y se forma un enolato. Este tipo de control sólo funciona si la diferencia de la acidez es lo suficientemente grande y no exceso de base se utiliza para la reacción. El control es más simple si sólo uno de los reactivos tiene protones ácidos y sólo esta molécula forma el enolato. Por ejemplo, la adición de malonato de dietilo en benzaldehído sólo produciría un producto:
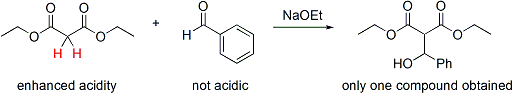
En este caso, el doblemente activados protones de metileno de la malonato se desprotona preferentemente por etóxido de sodio y cuantitativamente formar el enolato de sodio. Desde benzaldehído no tiene alfa-protones ácidos, sólo hay una posible combinación nucleófilo-electrófilo; por lo tanto, el control se ha logrado. Tenga en cuenta que este enfoque combina dos elementos de control de: aumento de la acidez de los protones alfa en el nucleófilo y la falta de protones alfa en el electrófilo.
El orden de adición
Una solución común es formar el enolato de una pareja en primer lugar, y luego añadir el otro socio en virtud control cinético. Control cinético significa que la reacción de adición aldólica hacia delante debe ser significativamente más rápido que la reacción retro-aldólica inversa. Para que este enfoque tenga éxito, otras dos condiciones también deben ser satisfechos; a saber, debe ser posible formar cuantitativamente el enolato de una pareja y la reacción aldólica hacia delante debe ser significativamente más rápido que la transferencia del enolato de una pareja a otra. Condiciones de control cinético comunes implican la formación del enolato de una cetona con LDA a -78 ° C, seguido por la adición lenta de un aldehído.
Enolatos
Formación
El enolato se puede formar mediante el uso de una base fuerte ("duras condiciones") o usando una Ácido de Lewis y una base débil ("condiciones suaves"):

Para desprotonación que se produzca, el requisito estereoelectrónico es que la alfa-CH enlace sigma debe ser capaz de superponerse con el pi * orbital de la carbonilo:
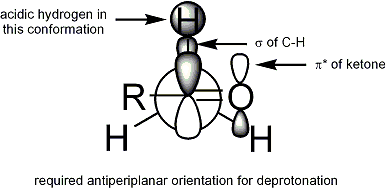
Geometría
Los numerosos estudios se han realizado sobre la formación de enolatos bajo muchas condiciones diferentes. Ahora es posible generar, en la mayoría de los casos, la geometría enolato deseada:

(- En la imagen de arriba, el segundo esquema de reacción debería decir> 99% E-enolato, no Z -) Para cetonas, la mayoría de las condiciones de enolización dan enolatos Z. Para ésteres, la mayoría de las condiciones de enolización dan enolatos E. La adición de HMPA se conoce para invertir la estereoselectividad de desprotonación.

La formación estereoselectiva de enolatos se ha racionalizado con el llamado modelo de Irlanda, aunque su validez es algo cuestionable. En la mayoría de los casos, no se sabe que, si las hay, son intermedios monomérico o oligomérica en la naturaleza; Sin embargo, el modelo de Irlanda sigue siendo una herramienta útil para la comprensión de enolatos.
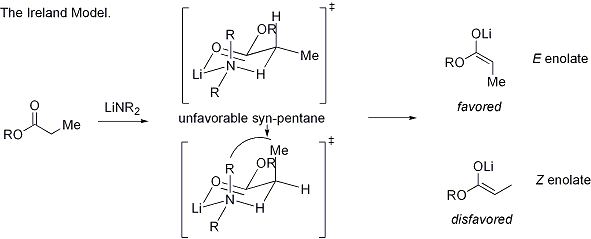
En el modelo de Irlanda, la desprotonación se supone que proceder por un estado de transición monomérico de seis miembros. El mayor de los dos sustituyentes en el electrófilo (en el caso anterior, metilo es más grande que de protones) adopta una disposición ecuatorial en el estado de transición favorecida, conduciendo a una preferencia por enolatos E. El modelo falla claramente en muchos casos; Por ejemplo, si la mezcla disolvente se cambió de THF al 23% HMPA-THF (como se ve arriba), la geometría enolato se invierte inexplicablemente.
Vs. Kinetic enolatos termodinámicas
Si una cetona asimétrica se somete a la base, que tiene el potencial para formar dos enolatos regioisómeros (ignorando la geometría enolato). Por ejemplo:

El enolato trisustituido se considera el enolato cinética mientras que el enolato tetrasustituido se considera el enolato termodinámico. El hidrógeno alfa desprotonado para formar el enolato cinética está menos impedido, y por lo tanto desprotonado más rápidamente. En general, las olefinas tetrasustituidos son más estables que las olefinas trisustituidas debido a la estabilización hiperconjugativo. La relación de regioisómeros enolato está fuertemente influenciado por la elección de la base. Para el ejemplo anterior, el control de cinética puede establecerse con LDA a -78 ° C, dando 99: 1 de selectividad cinética: enolato termodinámico, mientras que el control termodinámico puede establecerse con triphenylmethyllithium en temperatura ambiente, dando 10:90 selectividad.
En general, enolatos cinéticos se ven favorecidas por temperaturas frías, enlaces metal-oxígeno relativamente iónicos, y desprotonación rápida utilizando un ligero exceso de una base fuerte, mientras que obstaculizado enolatos termodinámicas se ven favorecidas por temperaturas más altas, enlaces metal-oxígeno relativamente covalentes, y de equilibrado ya veces para desprotonación usando una cantidad pequeña sub-estequiométrica de base fuerte. El uso de una cantidad sub-estequiométrica de base permite que una pequeña fracción de compuesto de carbonilo unenolized para equilibrar el enolato a la regioisómero termodinámico, al actuar como un servicio de transporte de protones.
Estereoselectividad
La reacción aldólica es particularmente útil porque dos nuevos centros estereogénicos se generan en una reacción. Una amplia investigación se ha realizado para entender el mecanismo de reacción y mejorar la selectividad observada bajo muchas condiciones diferentes. La convención syn / anti se utiliza comúnmente para denotar la estereoquímica relativa en la α- y β-carbono.

La convención se aplica cuando se añaden propionato (o de orden superior) nucleófilos a aldehídos. El grupo R de la cetona y el aldehído grupo de la R 'están alineados en un patrón "zig zag" en el plano del papel, y la disposición de los estereocentros formados se considera syn o anti, dependiendo si están en la misma o en los lados opuestos de la cadena principal.
Papeles viejos utilizan el eritro - treo nomenclatura familiar de química de carbohidratos.
E vs Z enolatos
No hay ninguna diferencia significativa entre el nivel de estereoinducción observado con enolatos E y Z:
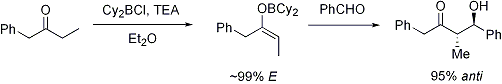

Ion de metal
El catión de metal enolato puede jugar un papel importante en la determinación del nivel de estereoselectividad en la reacción aldólica. El boro se utiliza a menudo porque su longitudes de enlace son significativamente más corto que el de otros metales tales como litio , aluminio , o magnesio . Por ejemplo, los bonos de boro-carbono y boro-oxígeno son 01.04 a 01.05 Å y 1.5 a 1.6 Å de longitud, respectivamente, mientras que típico enlaces metal-oxígeno-carbono y de metal son típicamente 1.9 a 2.2 Å y 2,0-2,2 Å de longitud, respectivamente. Esto tiene el efecto de "apretar" la estado de transición:

Estereoselectividad: Alfa estereocentro en el enolato
La reacción aldólica puede exhibir "estereocontrol basados en sustrato", en la que existente quiralidad en cualquiera de los reactivos influye en el resultado estereoquímico de la reacción. Esto ha sido ampliamente estudiado, y en muchos casos, puede predecir el sentido de inducción asimétrica, si no el nivel absoluto de diastereoselectividad. Si el enolato contiene una estereocentro en la posición alfa, excelente estereocontrol puede ser realizado.

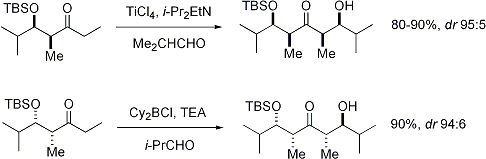
En el caso de un enolato E, el elemento de control dominante es alílico 1,3-deformación mientras que en el caso de un enolato Z, el elemento de control dominante es la evitación de interacciones 1,3-diaxiales. El modelo general se presenta a continuación:
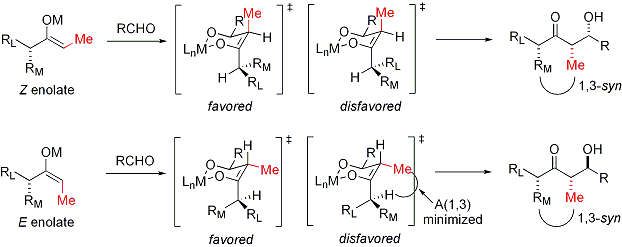
Para mayor claridad, el estereocentro en el enolato ha sido epimerizado; en realidad, el diastereoface opuesto del aldehído habría sido atacado. En ambos casos, el diastereómero 1,3-syn se ve favorecida. Hay muchos ejemplos de este tipo de estereocontrol:
Estereoselectividad: Alfa estereocentro en el electrófilo
Cuando enolatos ataques de aldehídos con un estereocentro alfa, excelente estereocontrol también es posible. La observación general es que E enolatos exhibición Felkin selección diastereoface, mientras que Z enolatos exhiben anti-Felkin selectividad. El modelo general se presenta a continuación:
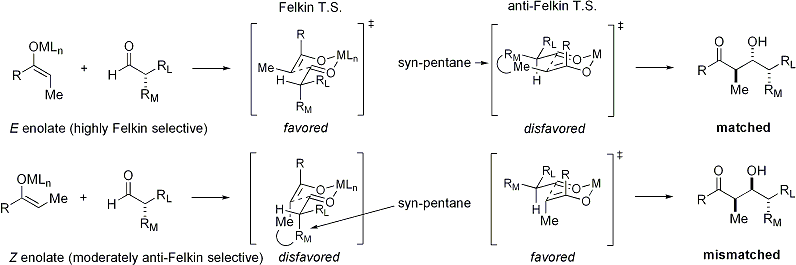
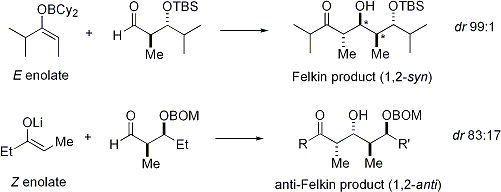
Desde Z enolatos deben reaccionar a través de una estado de transición que, o bien contiene una interacción syn-pentano desestabilizador o anti-Felkin rotámero, Z-enolatos exhiben menores niveles de diastereoselectividad en este caso. Algunos ejemplos se presentan a continuación:
Estereoselectividad: modelo fusionado para estereoinducción
Si tanto el enolato y el aldehído ambos contienen quiralidad pre-existente, entonces el resultado de la "doble stereodifferentiating" aldol reacción puede predecirse utilizando un modelo estereoquímica fusionado que tiene en cuenta el sesgo enolato facial, geometría enolato, y el aldehído sesgo facial. Se presentan varios ejemplos de la aplicación de este modelo a continuación: 
La química de oxazolidinona de Evans
Síntesis orgánicas modernas ahora requieren la síntesis de compuestos en forma enantiopuro. Puesto que la reacción de adición aldólica crea dos nuevos estereocentros, hasta cuatro estereoisómeros puede resultar.

Muchos métodos que controlan tanto la estereoquímica relativa (es decir, syn o anti, como se discutió anteriormente) y absoluta estereoquímica (es decir, R o S) se han desarrollado.
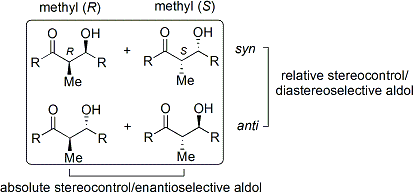
Un método muy utilizado es el de Evans acilo método oxazolidinona. Desarrollado en la década de 1970 y 1980 por David A. Evans y compañeros de trabajo, el método funciona mediante la creación temporalmente un enolato quiral añadiendo un auxiliar quiral. La quiralidad pre-existente de la auxiliar se transfiere entonces al aducto de aldol mediante la realización de una reacción aldólica diastereoselectiva. Tras la posterior eliminación del auxiliar, el estereoisómero deseado aldol se revela.
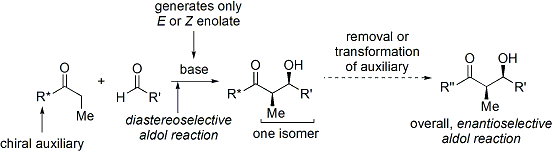
En el caso del método de Evans, el auxiliar quiral es un adjunto oxazolidinona, y el compuesto de carbonilo resultante es una imida. Un número de oxazolidinonas son ahora fácilmente disponibles en ambas formas enantiómeras. Estos pueden costar unos $ 10- $ 20 dólares por gramo, haciéndolos relativamente caro.
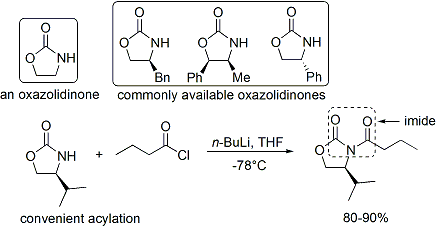
La acilación de una oxazolidinona es un procedimiento conveniente, y se conoce informalmente como "carga de hecho". Z-enolatos, que conducen a aductos syn-aldol, se pueden formar de manera fiable utilizando enolización suave de boro mediada:
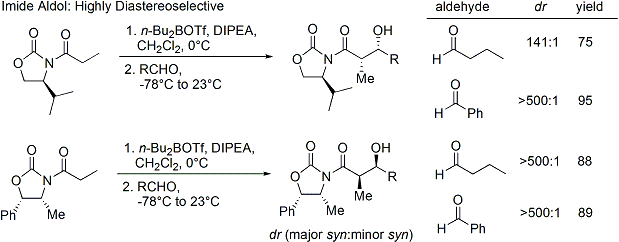
A menudo, un solo diastereoisómero puede obtenerse por uno la cristalización del aducto de aldol. Desafortunadamente, aductos de anti-aldólicas no se pueden obtener de forma fiable con el método de Evans. A pesar del costo y la limitación para dar sólo aductos syn, fiabilidad superior del método, facilidad de uso y versatilidad hacen que el método de elección en muchas situaciones. Muchos métodos están disponibles para la escisión del auxiliar:

Tras la construcción de la imida, tanto las reacciones de adición aldólica syn y anti-selectivas se pueden realizar, permitiendo que el conjunto de tres de los cuatro posibles stereoarrays: selectiva syn: y anti selectiva:

En las reacciones syn-selectiva, ambos métodos enolización dan la Z enolato, como se esperaba; sin embargo, el resultado estereoquímico de la reacción está controlada por el estereocentro metilo, en lugar de la quiralidad de la oxazolidinona. Los métodos descritos permiten que el conjunto estereoselectiva de policétidos, una clase de productos naturales que a menudo cuentan con el Retron aldólica.
Química Aldol Moderno
Metodología reciente permite ahora a una variedad mucho más amplia de reacciones aldólicas se lleve a cabo, a menudo con una cantidad catalítica de ligando quiral. Cuando las reacciones emplean pequeñas cantidades de enantioméricamente puros ligandos para inducir la formación de productos enantioméricamente puros, las reacciones se denominan normalmente "catalítico, asimétrica"; por ejemplo, muchos catalítico diferente, reacciones aldólicas asimétricas ya están disponibles.
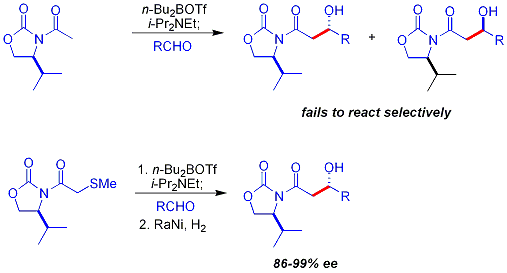
Reacciones Acetato Aldol
Una limitación clave de la enfoque auxiliar quiral descrito anteriormente es el fracaso de N-acetil imidas para reaccionar selectivamente. Un enfoque inicial fue usar un temporal grupo tioéter:
Reacción aldólica Mukaiyama
La Mukaiyama reacción aldólica es la adición nucleofílica de enol éteres de sililo a aldehídos catalizada por una Ácido de Lewis tal como trifluoruro de boro o cloruro de titanio. La reacción aldólica Mukaiyama no sigue el modelo de Zimmerman-Traxler. Carreira ha descrito metodología asimétrica particularmente útil con acetales de cetena sililo, destaca por sus altos niveles de enantioselectividad y alcance sustrato de ancho.
El método funciona en aldehídos alifáticos no ramificados, que a menudo son pobres electrófilos para los procesos catalíticos, asimétrica. Esto puede ser debido a la mala diferenciación electrónica y estérica entre su enantiofaces.

El análogo viníloga proceso aldólica Mukaiyama también se puede representar catalítico y asimétrica. El ejemplo que se muestra a continuación funciona de manera eficiente para aldehídos aromáticos (pero no alifáticos) y se cree que el mecanismo para involucrar a, dienolato unido al metal quiral.
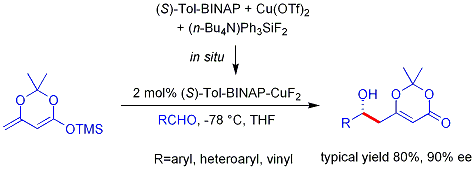
Crimmins thiazoldinethione aldólica
Una versión más reciente del auxiliar de Evans es el thiazoldinethione Crimmins. La rendimientos, diastereoselectivities, y enantioselectividades de la reacción son generalmente altos, aunque no tan alta como en los casos Evans comparables. A diferencia de la auxiliar de Evans, sin embargo, el thiazoldinethione puede realizar reacciones aldólicas de etilo (ref:... Crimmins, Org Lett 2007, 9 (1), 149-152) y puede producir el "syn Evans" o "no-Evans syn" aductos simplemente variando la cantidad de (-) - Esparteína. Se cree que la reacción proceda a través de seis miembros, titanio-bound estados de transición, análogas a los estados de transición propuestos para el auxiliar de Evans.
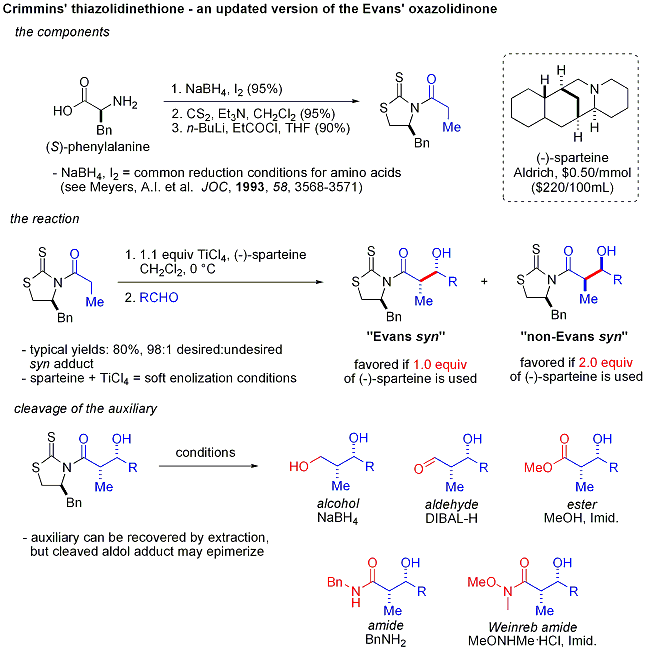
Reacciones aldólicas organocatalíticas
Un nuevo y emocionante desarrollo es el uso de secundarias quirales amina catalizadores. Estas aminas secundarias forman transitoria enaminas cuando se expone a las cetonas, que pueden reaccionar con electrófilos enantioselectiva aldehído adecuado. Esto se conoce como la catálisis enamina, un tipo de organocatálisis, ya que el catalizador se basa totalmente en una pequeña molécula orgánica. En un ejemplo seminal, prolina catalizada de manera eficiente la ciclación de un tricetona:

Esta reacción se conoce como la reacción de Hajos-Parrish (también conocido como la reacción de Hajos-Parrish-Eder-Sauer-Wiechert, refiriéndose a un informe contemporánea de Schering de la reacción bajo condiciones más severas). Sólo es necesaria una cantidad catalítica de prolina (3 mol%). No hay peligro de una reacción fondo aquiral debido a que los intermedios de enamina transitorios son mucho más nucleófilo que sus enoles de cetona padre. Esta estrategia es especialmente potente, ya que ofrece una forma sencilla de generar enantioselectividad en reacciones sin utilizar metales de transición, que tienen las posibles desventajas de ser tóxicos o caros.
Curiosamente, las reacciones aldólicas catalizadas por prolina no muestran efectos no lineales (la enantioselectividad de los productos es directamente proporcional a la enantiopureza del catalizador). Combinado con pruebas marcaje isotópico y estudios computacionales , la propuesta mecanismo de reacción para reacciones aldólicas catalizadas por prolina es el siguiente:

Esta estrategia permite que la reacción aldólica cruzada de otra manera desafiante entre dos aldehídos. En general, las reacciones cruzadas aldol entre aldehídos son típicamente un reto, ya que pueden polimerizar fácilmente o reaccionar no selectivamente para dar una mezcla estadística de productos. El primer ejemplo se muestra a continuación:

En contraste con la preferencia por aductos syn observados típicamente en adiciones aldólicas basado en enolato, estas adiciones aldólicas organocatalyzed son anti-selectivo. En muchos casos, las condiciones organocatalíticas son lo suficientemente suaves para evitar la polimerización. Sin embargo, la selectividad requiere la adición lenta jeringa bomba controlada del compañero electrofílico deseado debido a que ambos socios reaccionan normalmente tienen protones enolizables. Si un aldehído no tiene protones enolizables o alfa o beta-ramificación, control adicional puede ser alcanzado.
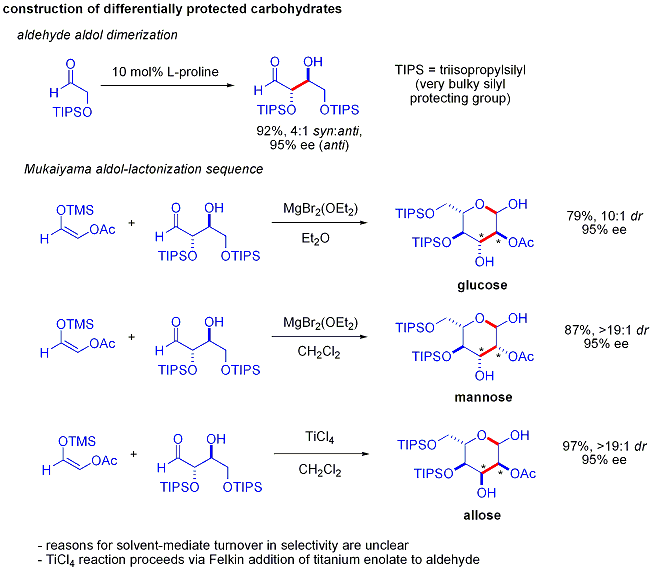
Un elegante demostración del poder de las reacciones aldólicas organocatalíticas asimétricas se dio a conocer por MacMillan y colaboradores en 2004 en su síntesis de protegidos de forma diferencial carbohidratos . Mientras que los métodos sintéticos tradicionales lograr la síntesis de hexosas utilizando variaciones de la iterativo las estrategias de protección-desprotección, requiriendo 8-14 pasos, organocatálisis pueden acceder a muchos de los mismos sustratos utilizando un eficiente protocolo de dos etapas que implica la dimerización catalizada por prolina de alfa-oxyaldehydes seguido por ciclación en tándem Mukaiyama aldol.
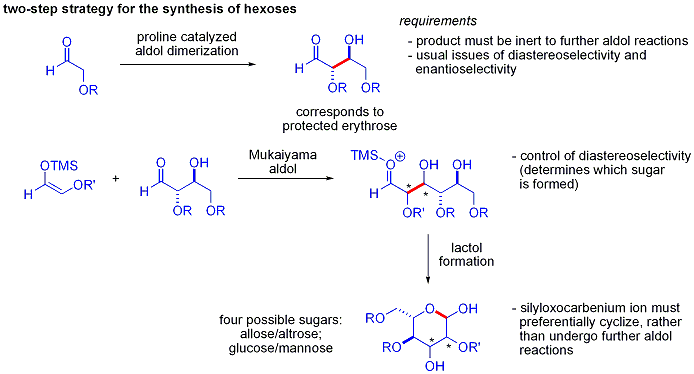
La dimerización aldólica de alfa-oxyaldehydes requiere que el aducto de aldol, por sí misma un aldehído, sea inerte para promover reacciones aldólicas. Estudios anteriores revelaron que los aldehídos de cojinete alfa-alquiloxi o alfa- sililoxi sustituyentes eran adecuados para esta reacción, mientras que los aldehídos teniendo Grupos aceptores de electrones tales como acetoxi eran no reactivo. El protegido producto eritrosa podría entonces ser convertida a cuatro azúcares posibles a través de Mukaiyama adición aldólica seguido de formación lactol. Esto requiere diastereocontrol apropiado en la adición aldólica Mukaiyama y el producto ion silyloxycarbenium a preferencialmente cicla, en lugar de someterse a una reacción aldólica. Al final, la glucosa , manosa, y alosa se sintetizaron:
adiciones aldólicas "directos"
En la adición usual aldólica, un compuesto de carbonilo se desprotona para formar el enolato. El enolato se añade a un aldehído o cetona, que forma un alcóxido, que se protona a continuación en estudio diagnóstico. Un método superior, en principio, se evitaría la secuencia de desprotonación-aldol-protonación en favor de una "adición aldólica directa". El principal problema en dicho proceso es que la adición aldólica genera un alcóxido, que es mucho más básico que los materiales de partida, lo que impide rotación de catalizador:

Un enfoque, se ha demostrado recientemente por Evans, es sililar el aducto de aldol:

Este método es más rentable e industrialmente útil que los procedimientos más típicos a base de enolato. Una más reciente, el enfoque biomimético por Shair utiliza beta-thioketoacids como nucleófilo. El resto es cetoácido descarboxilado in situ (la ligando quiral es una bisoxazolina). Curiosamente, aldehídos alifáticos y aromáticos ramificados son típicamente sustratos pobres.
